
Porphyrin versus Pyrrole Testing. Is there a Difference?
Annalies Corse BMedSc, BHSc
Pyrroluria is a condition known by many different names including Pyrrole Disorder, Mauve Factor and the older, unused names Kryptopyrroluria and Malvaria. It is described as being a metabolic condition, which essentially means it is a condition of altered body biochemistry. Pyrroluria disrupts a very specific aspect of our human biochemistry involving haem, an important chemical component of haemoglobin (the major oxygen carrying protein found in our red blood cells). It is also described as a genetic condition, with mounting clinical evidence the condition can be passed on from one generation to the next. Whilst a gene for Pyrroluria is yet to be found, the condition runs strongly through families. Experts in this field estimate that if a parent has Pyrroluria, there is a 50:50 chance it will be passed on to a child. This rises to a 75% chance of inheriting the disorder if both parents have it.
The name Pyrroluria is a technical term that essentially means excess levels of pyrroles in the urine. The condition was initially identified in the 1950’s and was known as ‘Malvaria’. Pioneers in the field of Pyrroluria research include Dr Abram Hoffer, Dr Humphrey Osmond and Dr Carl Pfeiffer. The condition is recognized by Doctors of Orthomolecular Medicine, Orthomolecular Psychiatry and Integrative Medicine, but remains largely unknown in Orthodox Medicine. Patients with Pyrroluria will also have problems associated with deficiencies of vitamin B6 and zinc. This is because the chemical by-product associated with Pyrroluria binds to both B6 and zinc, reducing the levels of the nutrients available for important biochemical pathways. Signs and symptoms of pyrroluria are essentially those of B6 and zinc deficiency and can include:
- Anxiety
- Mood swings, outbursts of temper, depression
- Sensitivity to loud noises and bright lights
- Morning nausea and poor morning appetite
- Histrionic (dramatic)
A second, less subtle condition related to Pyrroluria is known as Porphyria disorder. This is a group of disorders with abnormalities involving the synthesis of haeme. These are again genetic in origin, and mainly affect the skin (cutaneous porphyria’s) and the nervous system (cutaneous porphyria’s). The prevalence around the world varies dramatically, with estimates ranging from 1 in 500 to 1 in 50, 000 worldwide having a genetic trait that causes some form of porphyria. The condition is rare and has extremely varied presentations, ranging from severe anaemia’s, severe cutaneous blistering and serious digestive disturbances with seizures. Acute attacks often present during times of physiological stress.
For parents and carers (and indeed, practitioners), finding useful information regarding Pyrolluria can be a very lengthy and frustrating experience. Much of the information available online is from commercial sources or may not be written by scientifically qualified health professionals. The good quality, scientifically based information is out there, but it can be difficult to understand the scientific terminology used.
To add further confusion to this condition, there are currently two methods of laboratory assessment for Pyrrole Disorder; testing the urine for hydroxyhemopyrrolin-2-one (HPL), the key structural component of Mauve Factor. This is the test that has been honed and developed with specificity for Pyrolluria testing in mind. Additionally, porphyrin testing is occasionally suggested as another laboratory option when investigations for Pyrrole Disorder are warranted.
Porphyrin Testing
We all make porphyrins; they are essential structures required for the synthesis of haemoglobin and the cytochromes of energy production and liver detoxification pathways (to name a few). Naturally, we also must excrete porphyrins to prevent their build up. In orthodox medicine, a group of very rare diseases known as the Porphyrias involves the incorrect breakdown and excretion of haemoglobin. They are present is very large amounts in those with Porphyria and can be detected in blood, urine and fecal samples. The porphyrias are readily diagnosed by laboratory testing, especially at or near the time of symptoms. However, the number of tests available is very large, and the results among laboratories are not always reliable. For these reasons, it is important to locate a laboratory skilled in performing tests for Porphyria and a physician skilled in interpreting the test results. At present, some laboratories offer porphyrin testing as an alternative to pyrrole testing. Both the porphyrin structures and pyrrole structures are breakdown products of haemoglobin; porphyrins can simply be broken down further into pyrroles. Arguments in favor of porphyrin testing are that the structures tested are relatively stable, though it is not specific for Pyrrole Disorder. With pyrrole testing, if followed correctly it is highly specific for Pyrrole Disorder, but the substance tested is very fragile and prone to breakdown if not handled correctly by patients and lab staff.
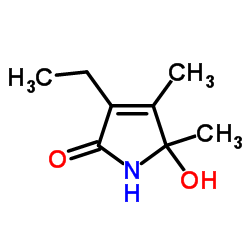
Figure 1: Hydroxyhemopyrrolin-2-one (HPL). Image source: Chemspider
Pyrrole Testing
Reliable pathology testing for Pyrroluria is available both in Australia and overseas. The testing method is based on urine analysis and determines if the patients urine a) tests positive for the presence of excessive amounts pyrroles and b) determines (quantifies) the actual level present in the urine. Pathology testing is essential for both adults and children with Pyrroluria, and correct diagnosis is essential to rule out other possible conditions that can often present in the same way.
Laboratory research into Pyrroluria has been occurring since the early 1970’s, with pathology testing becoming more sensitive and specific in that time. Original research thought that Mauve Factor was a chemical compound known as kryptopyrrole. However, as technology has improved it has been identified that a related compound, hydroxyhemopyrrolin-2-one (HPL) is the key structural component of Mauve Factor. Laboratories will usually be looking to detect HPL in their testing methods. Pyrroles are excreted from the body in urine, so this test is a reliable marker for detection of this metabolic condition.
This urine test involves the patient providing a small sample of urine, usually between 25ml to 50ml (the standard amount you would provide for most routine pathology testing). The normal amount of HPL in urine is less than 10mcg (micrograms) per dl (decilitre), often written as < 10mcg/dl on testing reports. To put this in context; there are 1000 micrograms in a milligram, and 10 decilitres in a litter, so what is being detected is a very tiny amount, yet significant enough to confirm the condition is present.
Urine must be collected in two vials, one of which contains ascorbic acid
(vitamin C). Both vials must be wrapped in foil and frozen immediately for transportation to the lab. The reason for foil wrapping and freezing is that pyrroles break down quickly and are highly unstable when exposed to light. Pyrroles are also very sensitive to oxidation by air, which is another reason why samples need to be handled with care and that all instructions regarding the specimen are followed correctly.
Once at the laboratory, the urine sample is tested via a process known as spectrophotometry. Spectrophotometry is used extensively in pathology and research laboratories all over the world for many types of tests besides Pyrroluria testing. It is a highly evidence based and reliable scientific technique when the detection of very small amounts of a substance is required. It is a relatively rapid method, where a machine known as a spectrophotometer measures the amount of light absorbed by the patients urine sample. Pyrroles absorb and reflect light at specific wavelengths, so their presence in urine can be detected by the spectrophotometer. Urine testing for Pyrroluria is quantitative, meaning that the quantity of pyrroles present can be measured as a numerical value (the intensity of the colour is recorded and the calculation of the result is carried out). So, rather than just receiving a result that states a patient is ‘positive’ or ‘negative’ for Mauve Factor, the test provides a value result. Quantitative pathology tests are very useful in this instance, as you can observe the level of pyrroles in the urine coming down with treatment, even though the level might still remain high. Practitioners and patients can use the test to observe the response to treatment, or if the treatment needs to be modified in some way. Seeing the pyrrole result decrease is very useful for patient compliance, even for children.
Test results that will require follow-up and possible referral and treatment include:
Borderline Pyrroluria: HPL between 10-20 mcg/dl
Positive Pyrroluria: HPL > 20 mcg/dl
In Australia, it is important to note if a laboratory is accredited with NATA for pathology testing. NATA is Australia’s authority responsible for the accreditation of laboratories, ensuring high standards of technical competence and best practice. If a lab is not accredited with NATA, you can discuss this with your practitioner, as they will have knowledge of the laboratories methods and quality control practices.
In addition to the urine test for Pyrroluria or porphyrins, a practitioner may suggest other tests at your initial appointment. Depending on the individual case and the symptoms present, it may also be necessary to have other functional or routing pathology testing organized, such as testing for methylation disorders or testing digestive health and bowel microbes. Testing can be expensive and it’s impossible to interpret the results and self-prescribe without clinical expertise, so ensure you have organized an appointment with an Integrative doctor first. It may be the case that only very few tests need to be organized when you consult with a health professional with experience in testing for and treating Pyrroluria.
Individuals with Pyrrole disorder require supplementation with B6, zinc and possibly evening primrose oil. This nutrient regime is essential to replace the nutrients that are rendered inactive by the build-up of HPL in the body. What is interesting is that each individual will require their own unique dose to achieve the best results, and the nutrients can be titrated (i.e., adjusted quickly) to deal with changing signs and symptoms, or increased during times of physiological stress to prevent attacks. This is most definitely a health condition that requires nutrient prescription from an integrative health practitioner.
Detecting Pyrroluria in adults is always a little trickier in clinical practice. Adults need to be willing to get tested, and willing to do the treatment. This is another reason why detecting the condition as early as possible in childhood is always the best option.
References
- Badawy A, Morgan, C. (1980). Tryptophan pyrrolase in haem regulation. The relationship between the depletion of rat liver tryptophan pyrrolase haem and the enhancement of 5- aminolaevulinate synthase activity by 2-allyl-2- isopropylacetamide. Biochemical Journal. 186(3): 763-72
- Harris K. Walsh Research Institute. BioBalance Conference. Pyrroluria, Gold Coast Conference; seminar given 2013 April 17.
- McGinnis W, Audhya T, Walsh W, Jackson J, McLaren-Howard J, Lewis A, et al. (2008). Discerning the Mauve Factor, Part 1. Alternative Therapies in Health and Medicine. 14 (2): 40-50
- McGinnis W, Audhya T, Walsh W, Jackson J, McLaren-Howard J, Lewis A, et al. (2008). Discerning the Mauve Factor, Part 2. Alternative Therapies in Health and Medicine. 14 (3): 56-62